FoldARE - overview
FoldARE is a computatonal tool for the prediction and analysis of RNA secondary structure.
The prediction step is based on a two-step strategy:
step 1 - from an RNA fasta:
(a) predicts an ensemble,
(b) through stastical weighting creates a pseudo-SHAPE co-input
step 2 - from the same RNA fasta, uses the pseudo-SHAPE co-input to predict the final 2D structure
It builds on the combination of some available, well established, methods: V, R, L, E (for ViennaRNA*, RNAstructure**, LinearFold, EternaFold). Any of these methods can be used interchangeably for creating ensembles (step1) or predicting w/ the pseudo-SHAPE co-input (step2).
* ViennaRNA/RNAfold is used in step2, ViennaRNA/RNAsubopt is used in step1 ** RNAstructure/Fold is used both in step1 and step 2
In its default mode, it runs EternaFold (the "ensembler", e; ensemble size = 75) for step1 and RNAstructure (the "predictor", p) for step2. In our benchmarks, this combination performed best. To change it, just select one of the available choices from the drop down-menu (for both p and e parameters) in Home page.
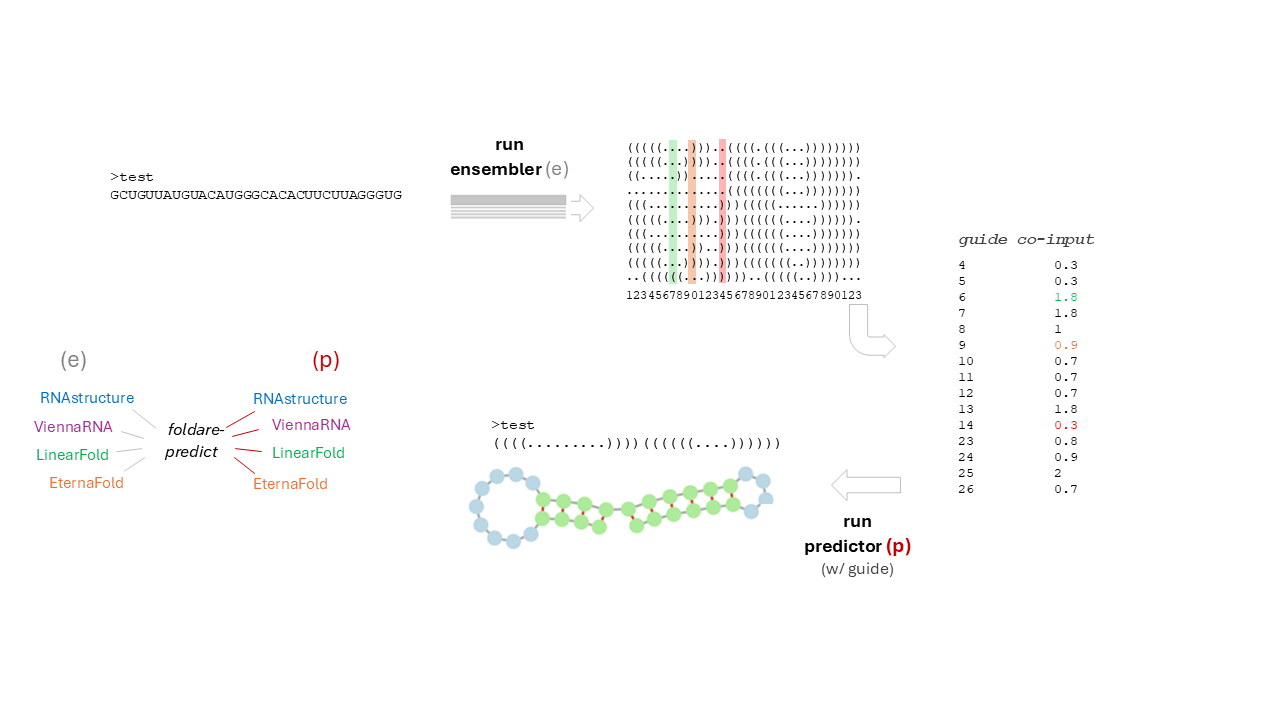
The core of the algorithm is the creation of a pseudo-SHAPE co-input file: it uses the ensemble data from step1 to derive (i) the frequencies per position of single strand predictions (e.g. 0-1 normalized frequency of "." per position, see Figure, shaded columns), and then (ii) weights this frequency: if high (e.g. > 0.9) a maximum weight (e.g. 2.0), if middle range (e.g. 0.5 to 0.9%) a mid range weight (e.g. 1.0), if low (e.g. 0.3 to 0.5) a low weight (e.g. 0.5) is used as a multiplier for the frequency to derive a pseudo-SHAPE value, indicative of its tendency to single-strandness/flexibility (e.g. if the frequency in the i-th position is 0.95, then a 2.0 weight is applied, f*w = 0.95*2 = 1.9, so a value of 1.9 will be stored in the pseudo-SHAPE co-input file, to be used in the next step (step 2, prediction with pseudo-SHAPE as guide co-input)
FoldARE-predict: input & output
The FoldARE prediction app takes in input a sequence, in fasta format (limited in size -- see Home page; for unlimited input size, and for full customization, we refer to the standalone version -- https://github.com/TebaldiLab/FoldARE).
The FoldARE-predict app accepts customization of the following parameters:
-p : choice of predictor (default, RNAstructure)
-e : choice of ensembler (default, EternaFold)
--ens_n: : ensemble size (default, 75)
--shape: optional custom SHAPE data (if not provided, pseudo-SHAPE is auto-generated)
The results page includes:
(i) the complete ensemble generated by the ensembler in dot bracket notation (input_e_ens.db)
(ii) The FoldARE-predict best model (MFE, default), in dot bracket notation (input_e_p_final.db
(iii) The FoldARE-predict best model drawn structure (input_e_p_final.svg)
(iv) The pseudo-SHAPE file (input_shape.txt) - or custom SHAPE if provided
(v) A summary file, detailing search parameters (with file name and content of all outputs)
Finally, at the top of the results page: a link to Download (as zip) all generated output files is available.
The FoldARE prediction algorithm has been tested, optmized and validated with different datasets (ArchiveII, RNAstrand, bpRNA, filtered by redundancy). The algorithm is described in detail in this manuscript doi:10.64898/2026.03.04.709501
FoldARE is available as a standalone, customizable tool in github/FoldARE.